Синдром на Maroteaux-Lamy или мукополизахаридоза VI е рядко наследено заболяване, при което носителите имат следните характеристики:
- Къс ръст,
- деформации на лицето,
- къс врат,
- повтарящ се отит,
- заболявания на дихателните пътища,
- скелетни малформации и
- мускулна скованост.
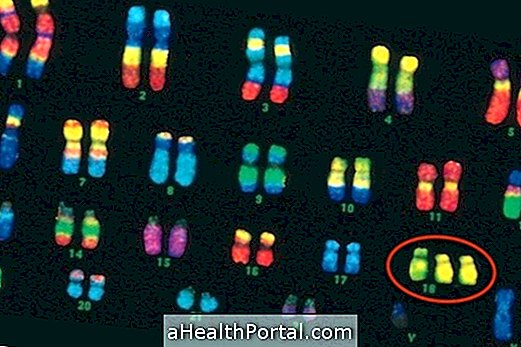
Заболяването се причинява от промени в ензима Арилсулфатаза В, което му пречи да изпълни своята функция, която да разгражда полизахаридите, които на свой ред се натрупват в клетките, развивайки симптомите, характерни за болестта.
Пациентите със синдрома имат нормална интелигентност, така че децата не се нуждаят от специално училище, само адаптирани материали, които улесняват взаимодействието с учителите и съучениците.
Диагнозата се прави от генетик, основаващ се на клинична оценка и лабораторни биохимични анализи. Диагнозата през първите години от живота е много важна за изготвянето на план за ранна намеса, който ще помогне за развитието на детето и за насочването на родителите към генетичното консултиране, тъй като те рискуват да преминат болестта към по-късните деца.
Няма лечение за синдрома на Maroteaux-Lamy, но някои лечения като трансплантация на костен мозък и ензимна заместителна терапия се оказват ефективни при намаляване на симптомите. Физическата терапия се използва за намаляване на сковаността на мускулите и увеличаване на движенията на тялото. Не всички страдащи имат всички симптоми на болестта, тежестта варира от индивид на индивид, някои са способни да живеят с относително нормален живот.