Фенилокетонурията е рядко генетично заболяване, характеризиращо се с наличието на мутация, отговорна за промяна на функцията на ензим в организма, отговорен за превръщането на аминокиселината фенилаланин в тирозин, което води до натрупване на фенилаланин в кръвта и което при високи концентрации е токсично за организъм, който може да причини интелектуални затруднения и конвулсии, например.
Това генетично заболяване има автозомно рецесивно естество, т.е. за да се роди детето с тази мутация, е необходимо двамата родители да са поне носители на мутацията. Диагнозата на фенилкетонурия може да се направи скоро след раждането чрез теста на крака и след това е възможно ранното лечение да се установи рано.
Фенилкетонурията няма лек, но лечението й е чрез храна и е необходимо да се избягва консумацията на храни, богати на фенилаланин, като например сирене и месо.

Основни симптоми
Новородените с фенилкетонурия първоначално не са имали симптоми, но симптомите се появяват няколко месеца по-късно, като основните са:
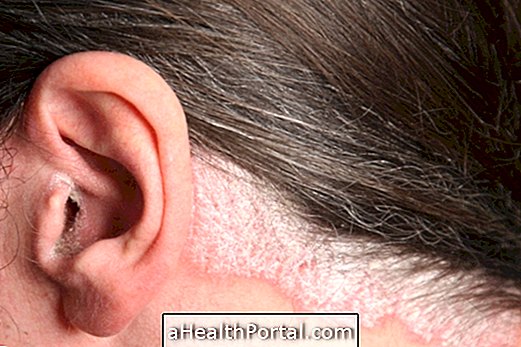
- Екзема-подобни рани на кожата;
- Неприятна миризма, характерна за натрупването на фенилаланин в кръвта;
- Гадене и повръщане;
- Агресивно поведение;
- хиперактивност;
- Умствено изоставане, обикновено тежко и необратимо;
- припадъци;
- Поведенчески и социални проблеми.
Обикновено тези симптоми се контролират чрез адекватна диета и ниско съдържание на фенилаланин. Освен това е важно лицето с фенилкетонурия да бъде редовно последвано от педиатър и диетолог от кърменето, така че да няма сериозни усложнения и да не се компрометира развитието на детето.
Как се извършва лечението?
Основната цел на лечението на фенилкетонурия е да се намали количеството фенилаланин в кръвта. Поради това обикновено се посочва приемането на диета с ниско съдържание на храни, съдържащи фенилаланин, като например храни от животински произход. Голяма част от храните, съдържащи фенилаланин, също са големи източници на желязо, така че добавката на желязо обикновено се посочва от вашия педиатър или специалист по хранене. Вижте кои храни са богати на фенилаланин.
Важно е човекът да бъде придружаван през целия живот и редовно, за да се избегнат усложнения, като например нарушения на централната нервна система. Научете как се извършва лечението на фенилкетонурия.
Жената с фенилкетонурия, която желае да забременее, трябва да бъде посъветвана от акушер-гинеколог и диетолог по отношение на рисковете от повишена концентрация на фенилаланин в кръвта. Поради това е важно редовно да се оценява от лекаря, в допълнение към диетата, подходяща за заболяването и вероятно допълване на някои хранителни вещества, така че майката и детето да са здрави. Научете как да контролирате фенилаланина по време на бременност.
Фенилкетонурията има лек?
Фенилкетонурията няма лек и следователно лечението се извършва само с контрол в диетата. Увреждането и интелектуалното увреждане, които могат да възникнат при консумирането на храни, богати на фенилаланин, са необратими при хора, които нямат ензим или имат нестабилен или неефективен ензим по отношение на превръщането на фенилаланин в тирозин. Тези щети обаче лесно могат да бъдат избегнати чрез хранене.

Как се прави диагнозата?
Диагнозата на фенилкетонурия се прави скоро след раждането чрез теста на крака, който трябва да се извърши между първите 48 и 72 часа от живота на бебето. Този тест е в състояние да диагностицира не само фенилкетонурия в бебето, но също и сърповидно-клетъчна анемия и муковисцидоза, например. Разберете кои са болестите, идентифицирани чрез теста на крака.
Децата, които не са диагностицирани чрез теста за краката, може да имат диагноза, извършена чрез лабораторни тестове за оценка на количеството фенилаланин в кръвта и в случай на много висока концентрация може да се извърши генетичен тест за идентифициране свързаната с болестта мутация.
Чрез молекулярни изследвания е възможно да се идентифицира ефектът на мутацията и по този начин да се определи тежестта на заболяването. Някои мутации са в състояние да намалят или забавят активността на ензима, докато други правят фенилаланина вече непризнат от ензима или превръщат ензима в много нестабилен и нормалното функциониране на неговата функция е трудно.
Определянето на вида на мутацията и концентрацията на фенилаланин в кръвта е много важно, така че лекарят да може да провери степента на заболяването и възможните усложнения, освен че помага на диетолога при изграждането на хранителния план, който винаги се основава на количеството фенилаланин в кръвта, Когато количеството е ниско, диетологът може да препоръча консумацията на храни, които съдържат малко количество фенилаланин в състава му и когато стойностите на тази аминокиселина са над идеалните, без фенилаланин храни.
Важно е, че дозата на фенилаланин в кръвта се прави редовно. В случая на бебета е важно да се прави всяка седмица, докато бебето завърши 1 година, докато за деца между 2 и 6 години изследването трябва да се извършва на всеки две седмици, а за деца от 7 години - месечно. Ранното диагностициране на фенилкетонурия позволява да се вземат необходимите грижи с храната от самото начало, като се избягват редица нежелани последствия, като трайно увреждане на мозъка.
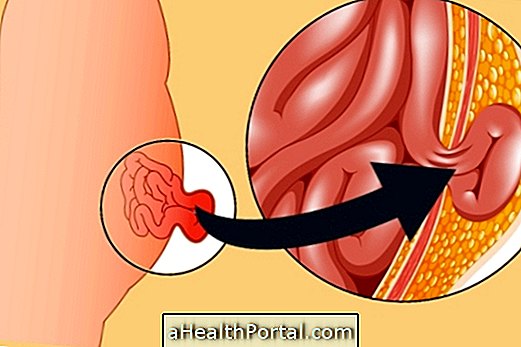
Тъй като фенилаланинът е токсичен за фенилкетонурика
Въпреки, че е налице в няколко храни, фенилаланинът трябва да се превърне в тирозин, което се дължи на действието на ензима фенилаланин хидроксилаза, наричана още ПАХ.
При хората с фенилкетонурия ензимът ПАХ е дефектирано произведен поради генетични промени, което означава, че няма превръщане на фенилаланин в тирозин и натрупване на фенилаланин в кръвта. Тази аминокиселина може също да се превърне в пирувинова киселина, което я прави доста токсичен за тялото и може да доведе до невронални промени.